Research Interests
Besides not being limited on, I am interested in parasitic Eukaryote pathogens genomics and the biology of genome evolution. How good the genomes available are? Do we have possible artifacts, such as compressed regions, misassembles, gaps, not optimized annotations and mixed population samples, used as reference by the community? How do genomes differ between populations and how do they evolve? Can we trace gene candidates for a specific phenotype or evolutionary significant? How about non-coding regions, could they tell something important about the organism? The answers to these questions are important because of the insight they provide into parasite biology, evolution and the identification of potential drug targets.
Eukaryotic pathogens are ideal for our studies for several reasons:
Our approach is to apply computational, phylogenetic and molecular approaches to analyze parasite genomes and its population. Projects include the development of tools for comparative genomics and assessing the phylogenetic distribution of coding (genes) and non-coding regions.
Eukaryotic pathogens are ideal for our studies for several reasons:
- They are relatively small. Most of their genomes are <100 Mb in length;
- The high number of species to be parasitic, which gives high importance for epidemiological and drug discovery health sciences studies;
- There are currently hundreds of genome sequences available for different species and thousands of new genome and strain sequencing data, including DNAseq (all kinds of platforms) and RNAseq being deposited in public databases;
- There are community databases, such as EuPathDB that work directly with the community to keep the most up-to-date sequence annotation available, interested in getting the best of these genomes.
Our approach is to apply computational, phylogenetic and molecular approaches to analyze parasite genomes and its population. Projects include the development of tools for comparative genomics and assessing the phylogenetic distribution of coding (genes) and non-coding regions.
Genomics |
|
Is reliance on an inaccurate genome sequence sabotaging your experiments?
Advances in genomics have made whole genome studies increasingly feasible across the life sciences. However, new technologies and algorithmic advances do not guarantee flawless genomic sequences or annotation. Bias, errors, and artifacts can enter at any stage of the process from library preparation to annotation. My goal is generate new data and revisit available genomes using all available "omic" data to detect and fix these artifacts to better understand de organism biology, evolution and give insights to experimental design helping the community to avoid a failed experiment or inconclusive result. |
|
Molecular Evolution & Phylogenomics

|
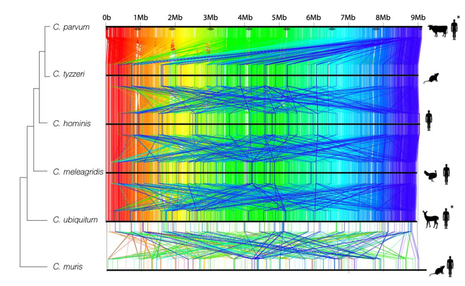
How do genomes differ between populations and how do they evolve?
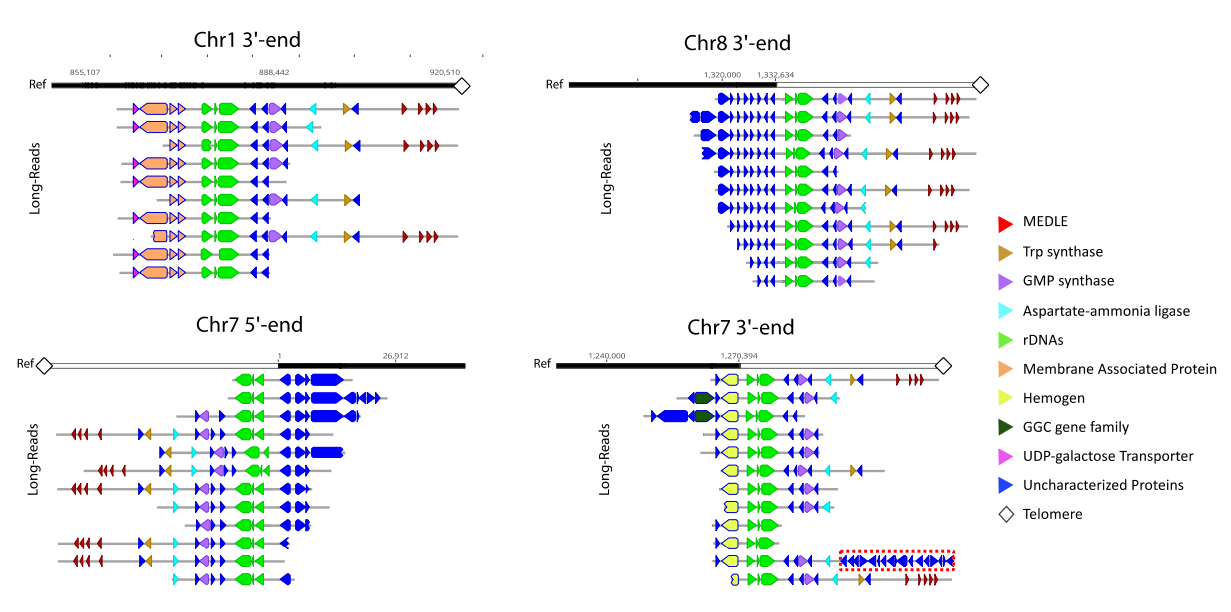
Inversions are potent forces in local adaptation and diversification. High-throughput genomics methods have revived the popularity of research on inversions, and recent studies reveal that they are taxonomically more widespread than previously thought. Inversions can be maintained over hundreds of thousands and even millions of years via various forms of selection and often involve large genomic regions, each comprising hundreds of genes, together representing a significant portion of the genome. My goal is to track these regions among populations and check their impact in the recombination rate and selection coefficient, which are usually biased by the current methods that do not take an account these inversions for their estimations. Can we trace gene gene candidates for a specific phenotype or evolutionary significant? In eukaryotes, a relatively high number of ORFs are identified that do not show a match with other known protein motifs in databases, usually known as "hypothetical proteins". These names came because of the assumption that their homologs are yet to be discovered. However, an alternative interpretation would be that some proteins evolve so fast that their homologs cannot be discovered over larger evolutionary distances. Tracing these genes in populations of same species could help to detect some of these fast evolving homologs and select gene sets that can tell precisely how the population is evolving and how to characterize them. Does mixed parasites populations clinically differ from monoclonal infections? Many field isolates from patients collected from outbreaks are known to be a result of multiple infections from parasites populations. Besides affecting some "omic" analysis, such as gene copy number variation, SNVs and genome assembly, these mixes increases the diversity of the infection which could cause different clinical manifestations. Our goal is to detect these populations in these mixed samples and verify if these are interacting between each other. |
Trancriptomics |
|
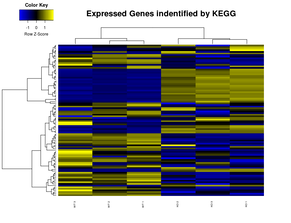
What differentiated expressed genes could tell about the parasite behavour?
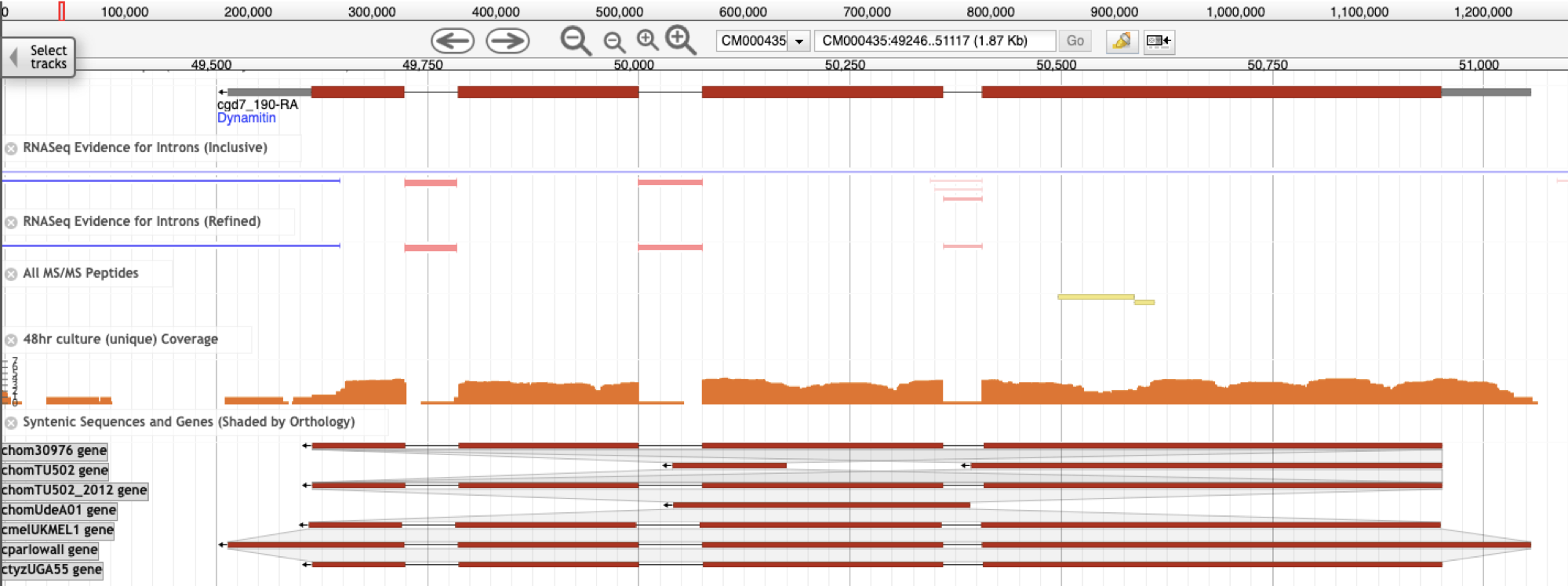
Different life cycle stages, treatments and/or different phenotypes could tell a lot how the organism is behaving in the expression level. Our goal is to detect differential expressed genes between these samples to better understand the parasite biology. What RNAseq data tell about the annotation? Many genes annotated in most genomes available are a result of "in silico" prediction tools and don't have any biological evidence for their structure. This lack of evidence makes some these genes to have a wrong/incomplete structure (missing introns, exons and UTR information) and also their potential isoforms. Our goal is to use transcriptomic data from both short and long read sequencing approaches to revisit some of these genome annotation. |
|
Metagenomics |
|
How the gut microbiome could affect the parasite development?
Many are the factors that a microbiome could interfere in the environment. Some eukaryotic pathogens, such as Apicomplexans are known to have endosymbiotic events and gene transfer during their evolutionary history. Some of the most important genes for some parasites survival are a fruit from these interactions. We believe that the microbiome could be directly interfering to the parasite evolution. We are more interested more specifically in Cryptosporidium infections in the gut. Our aim is to look how different microbiome environments (infected vs not infected or different diets) could affect the parasite development and genome content. |
